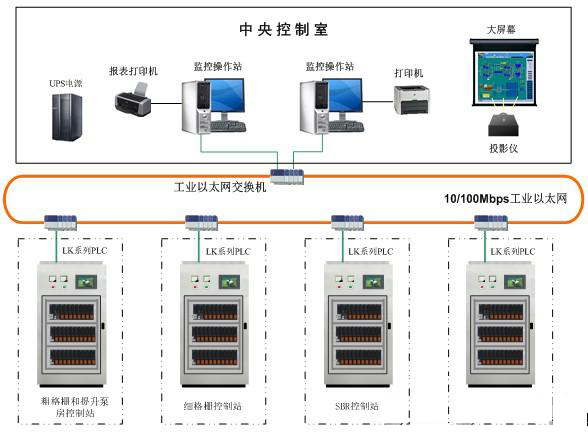
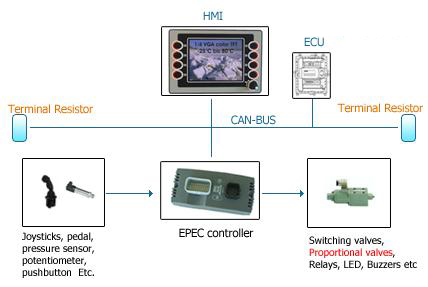
尽管在1997年3月已成为法律,最终用 户并不急于遵守美国食品和药品管理
局(FDA, Rockville, MD) 21 CFR有关电子记录和电子签名的Part 11 规程。用户常常引用 FDA 的需要为理由,为他们的放慢依从提供了相当充分的解释。
在2002 年4月出版的CONTROL ENGINEERING中发表了一篇题为“我从政府来,我会帮助你”的文章,总括了在电子记录和电子签名方面怎样达到符合 21 CFR 11规程和在实践过程中增进商业利益。
2002年11月,有关咨询更多细节的电话蜂拥而来,FDA发表一个题为“ 21 CFR Part 11,对食品和药品行业的指导:电子记录;电子签名;电子记录的电子拷贝”草案指导文件作为回答。
然后,2003年2月3日,FDA 宣布撤回它的 Pa
存在并很好发挥作用
真实情况是: FDA 并未做任何事去改变或影响 21 CFR Part 11 规程;它只是提出新的创议去增强其现行优良药品制造标准 (cGMP)规划,包括 Part 11。这只是听起来很简单。事实是在过去的25年中,许多 FDA 的优良药品制造标准(GMP)并没有被修改,因而对于希望应用新技术和新发明的用户来讲,FDA经常成了绊脚石。
依照其新的创举,FDA 试图:
◆资源和规程应集中注意力于那些对公众健康造成极大风险的制成品上;
◆ 确保 FDA的工作不会阻碍发明创造;
◆在其各个组成部分,强化 FDA规章方案的一致性。
作为所有改变的一部分,监督Part 11 的主要职责被转移到 FDA 的药品评估和研究中心 (CDER),它同样位于Rockville。
2003年2月20日,FDA 为21 CFR Part 11发表了一个新的草案指导文件,并提供60天的时间,公众可对此文件进行评论。

新的指导文件表明 FDA 打算重新审查 Part 11,可能对Part 11提出修改,作为它的“21世纪制药工业的 cGMPs的部分:一个基于风险的方案”创议的一部分。在FDA重新审查Part 11的同时,它打算狭义地解释Part 11的范围,并就特定的Part 11条例而言,应谨慎地实行强制性决定。
作为正常操作的部分,这意味着FDA将不采取行政措施来强制依从Part 11的有效性、审计跟踪,记录保存和记录拷贝等要求。FDA还表示对在Part 11的有效日期1997年3月以前运行的某些墨守成规的传统系统实施强制性决定会谨慎。
预测规则
新的指导草案强调预测规则要求的重要性,并需要文件化和说明风险评估的理由。此草案文件包括对软件和计算机系统上现有FDA 指导文件以及对国际药品工程师协会 (ISPE, Tampa, FL)的“对自动化系统有效性的指导”(GAMP)的某些引用。
就其狭义的解释,FDA 对仍然依从Part 11的记录将加强预测规则要求,因此,根据基本的预测规则和为了满足所有其他预测规则要求,被管理的行业应负责保持和提交安全和可靠的记录。
在此同时,FDA发表了它的新的指导文件,ISPE出版了题为“符合21 CFR Part 11的基于风险的方案”的指导文件。
基于风险的评估法则
以 Part 11为出发点,ISPE与 FDA合作,召集一组行业专家,采用基于风险的方案对所有的GMP记录调查其依从规程的程度。对该专家组的挑战是探索一种方法来回答 “它是一个GMP 记录吗?”而不是“它是一个电子记录吗?”这样的问题。
点击看原图
该组提交FDA评议的方法(该方法由FDA批准以便进一步拓展)提供:
◆ 范围— 用户企业应该在预测规则、过程的危险性、产品安全风险、有效性、一致性、纯度和/或质量的基础上,识别和定义有高度影响力的电子记录和电子签名。
◆选择— 用户企业必须实现与电子记录的临界性以及该记录识别的风险相当的控制。这些控制应文件化并有充分理由证明所识别的风险。
在CONTROL ENGINEERING对 Conformity公司 (Lianrwst, U.K.)董事兼ISPE技术顾问Sion Wyn的一次独家采访中,
某一个地区若在定量风险方面打算采用基于风险评估法,则可能会引发相近控制系统将最终产品抛售给公众从而导致混乱。表面上,似乎来自控制系统的最终产品越逆流而上,风险会越小,于是对有效性要求就不太会严格。但是,正如NuGenesis 技术公司(Westborough, MA)市场开发经理Victoria Landers指出的那样,风险取决于建立什么数据系统,如何使用数据以及数据的终点在何处。
Landers 女士说:“例如预-临床的毒理学测试,在药品发现和开发期间进行。如果这些测试完成得不好,或者对数据的解释错误,结果可能使最后的产品对公众的健康产生相反的效应。”
对 Part 11的影响
在最初提交给FDA的Part 11中,ISPE 委员会指出现行电子记录的定义范围太广,会潜在地窒息革新,并且使人们的注意力和资源脱离确保产品记录的完整性,这样做
从Part 11 形成法律起,电子记录的拷贝、保存和维护一直是热烈讨论的话题。在提议的新的Part 11导则下,用户应该能满足规程要求(按优先级排序):
◆只要利大于弊,应使用可移植的工业标准格式。
◆ 以包括PDF,microfiche(缩微胶片), microfile(缩微文件),或纸型拷贝等通常能接受的格式,使用常规的自动转换或输出方法来产生拷贝。
◆ 跟随厂商建立的步骤,使用厂商的硬件和现场软件,就能检验和审查记录。
委员会关于影响大的记录增加了一个申明,声称“对于影响大的记录,保留记录的电子拷贝也是必要的—虽然不一定要求是可重处理的形式。”
点击看原图
接近风险
分析风险是每天生活的一部分。
非正式的风险评估法则包括:在汽车逼近刚转为黄色的交通灯时决定踩油门还是刹车。
正式的、结构性的风险评估法则可应用到日常生活中的商业过程,如金融,市场,保险,气象和员工及社区安全。因此,不足为奇的就是FDA正在其本身资源中找寻合适的风险评估方法,来帮助获得它所陈述的21世纪药品cGMPs的创新:一种基于风险的方法。
FDA的发言人坚持任何用于被管理行业、基于风险的方法必须设计为防止不安全情况的发生。FDA坚决认为任何依赖于生产条件的现场检查和/或对最终产品的随机抽样来检测不安全条件的解决方法都是不合适的。
一个可能的风险评估候选方案是FDA食品安全和营养应用中心(CFSAN,College Park,MD)开发和使用的保证食品安全的FDA“危险性分析和临界控制点(HACCP)”指导文件。
CFSAN的HACCP指导文件应用了7个基本原则来满足其保证食品安全的目标:
◆分析潜在的危险和控制这些危险的手段;
◆ 确定临界控制点;
◆ 通过建立每个控制点的临界限制来建立预防措施;
◆ 建立监控临界控制点的方法;
◆ 当监控显示尚未达到临界限制时,建立须采用的纠正步骤;
◆ 建立确定系统是否在正常运行的检验步骤;
◆ 建立有效记录,坚持HACCP程序的文档化。
仅通过以上的简要说明,HACCP指导文件的流程和目的就很明显了。然而,诀窍在于弄清楚如何在系统中应用HACCP导则信息,以产生电子记录和电子签名。
有一种方法能识别高风险系统和低风险系统,然后集中资源识别高风险子系统。
有了系统和子系统优先等级表的武装,知识渊博的团队通过子系统的硬件和软件可能的表达方式来使用HACCP原则。
无疑,今年2月, FDA向21CFR Part11规程抛出了一个曲线球。 然而,一个细致的考核揭示所提议的更改,实际上将给公众提供更安全的产品,并使用户花费较少和更容易遵守,实现真正的双赢。
并且,在少数例外情况下,万一你对此有所怀疑,CONTROL ENGINEERING 2002年4月刊所提议的概念仍然有效。
相关更多信息,请
www.cechinamag/freeinfo输入咨询编号查询
Conformity
www.conform-it.com 255
FDA CDER cGMPs
www.fda.gov/cder/gmp/ 256
FDA CFSAN HACCP
www.cfsan.fda.gov/~lrd/haccp.html 257
ISPE
www.ispe.org 258
NuGenesis
www.21cfrpart11.com 259